
质粒纯化的最佳实践

质粒纯化是大多数分子生物学实验室中的常用技术。尽管标准的碱裂解方法已经很成熟,但仍然有很多事情可能出问题。本指南列出了质粒纯化的一些常见注意事项。
要做:使您的大肠杆菌变得丰富(至少是它们的培养基)
来自同一大肠杆菌原代菌株的质粒产量可能会因许多因素而有很大差异。LB(Lysogeny Broth)等标准培养基可以很好地生长和生产质粒。但是,更丰富的培养基(如Terrific Broth,“ TB”)可以提高每毫升培养物中质粒的产量。这对于增加从较小培养体积中收获的低拷贝质粒量特别有用。
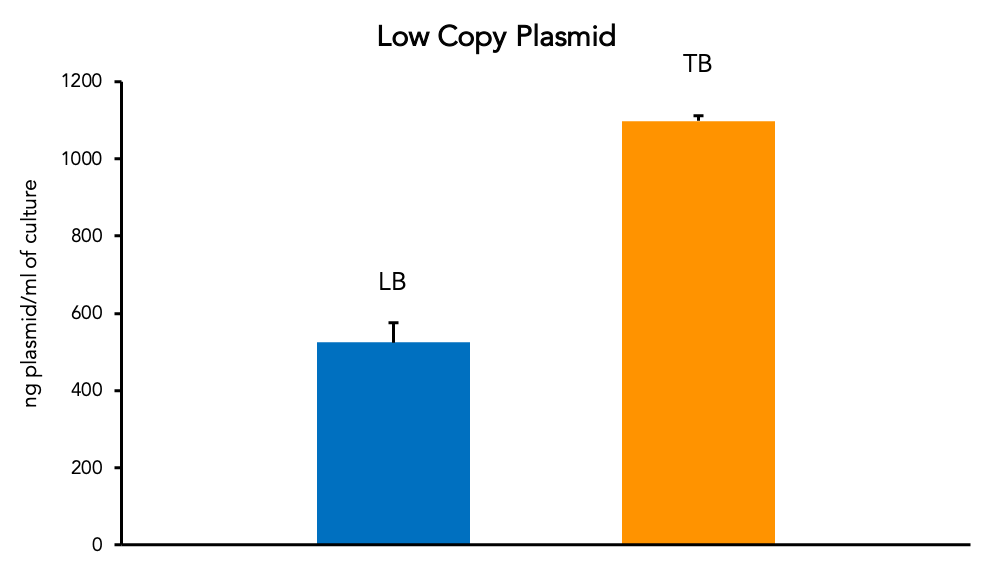
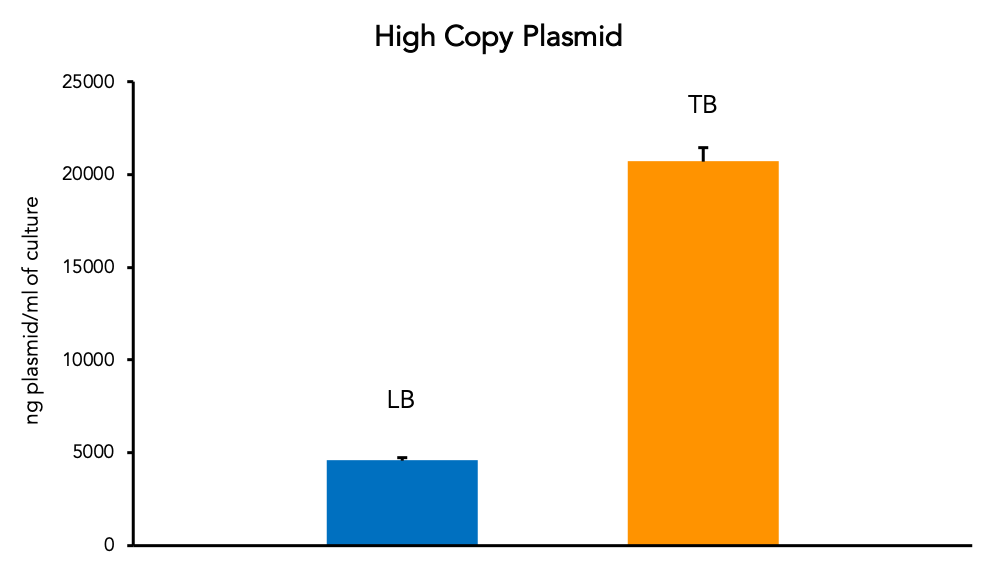
不要:忘了在培养基中添加抗生素
一个令人惊讶的常见错误,如果您忘记在培养物中添加抗生素(或添加得太少),则可能会由于选择压力不足而导致质粒丢失。确保仔细检查培养物中抗生素的浓度。
要做:摇匀
大肠杆菌培养通常在烧瓶中生长,需要适当的通气以获得最佳生长。通常,这需要在200-250 rpm(每分钟转数)下摇动培养物。此外,考虑通过使最小的培养物与空气体积比约为1:5来增加培养物通气量,并考虑使用带挡板的培养瓶。最后,请使用诸如铝箔(略松),棉塞或透氧膜之类的盖子确保培养物获得足够的通气。

不要:过度生长
如果不能使培养物生长太长时间,则更多的情况并没有改善,您会遇到基因组DNA污染和劣质质粒制备的情况。最好在接种后约12-18小时处理细胞,一旦培养物致密但在过度生长之前。
做:测量OD 600
OD 600是评估培养物密度的好方法,因此您知道何时处理细胞。由于无法测量高光密度,因此取培养液的等分试样,稀释十倍,然后使用标准分光光度计进行测量。培养物准备好以OD 600为0.2-0.35 进行处理以稀释十倍的培养物。
不要:超出培养限制
许多质粒制备试剂盒附带有关培养条件和输入的建议。请认真遵循这些步骤,以确保不会使色谱柱超载。添加过多的培养物是堵塞色谱柱并降低纯化性能的可靠方法。
做:温柔
正确纯化质粒的关键之一是从质粒DNA中分离基因组DNA。可以通过移液或涡旋将细菌沉淀重新悬浮在P1缓冲液中。但是,您不能在裂解过程中将其混合得太粗,因为您可能会剪切可与色谱柱基质结合并污染制备物的基因组DNA。
请勿:忽略裂解时间
某些人可能会认为,在裂解方面,越长越好,这是假定最好不要留下任何DNA。但是,大肠杆菌的碱裂解只能进行很长时间才能产生澄清的裂解物。更多的培养会导致更多的碎片,但是如果使用的裂解缓冲液太少,则可能没有额外的裂解或质粒释放。另外,不同菌株的大肠杆菌对碱裂解的敏感性不同。最重要的是,裂解时间过长可能会导致质粒DNA变性,从而导致制备质量不佳。
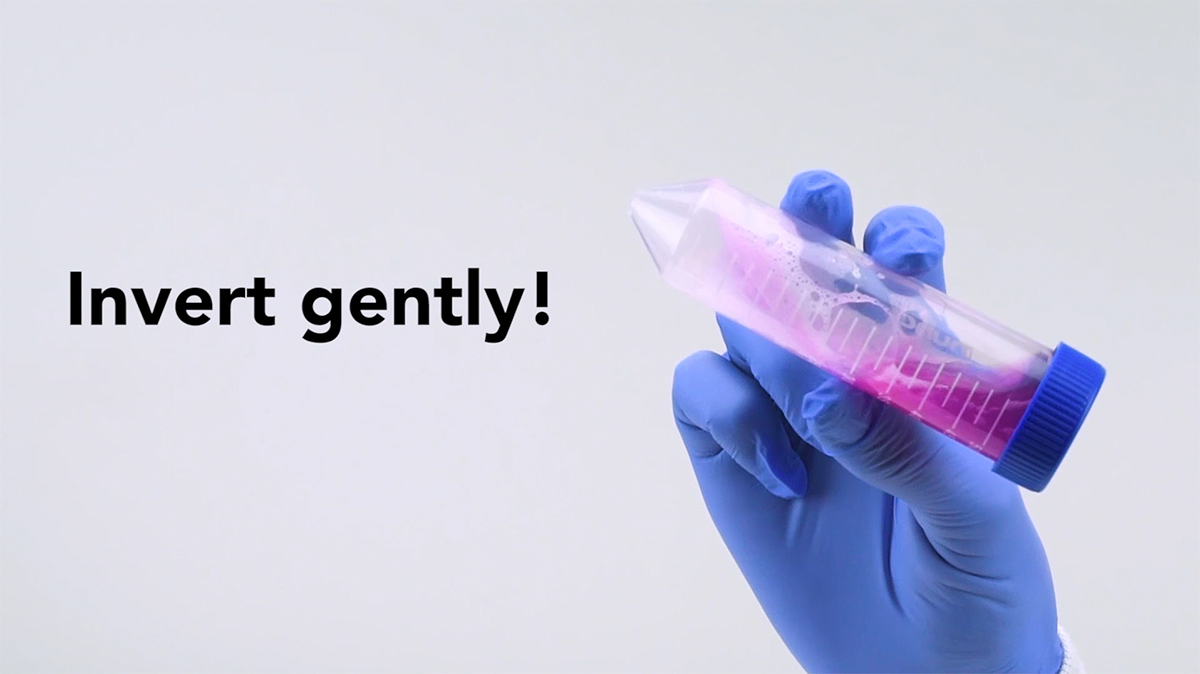
做:完全中和裂解液
添加中和溶液并与裂解液混合后,请勿立即进行下一步!中和不完全会导致制备质量差。确保多次颠倒试管,以确保裂解液被完全中和。即使观察到大量的蓬松沉淀物,也要多一点时间(请参阅规程)以确保溶液完全混合。

不要:吸取碎片
中和步骤后,许多质粒制备需要离心以从细胞碎片中分离出DNA,从而澄清了裂解液。在此步骤中,缓慢工作并避免将不必要的细胞碎片转移到可能干扰结合和/或降低纯度的下一步非常重要。
做:加酒精
许多质粒制备试剂盒包含使用乙醇的洗涤缓冲液。这些缓冲液通常是浓缩液,使用前必须在乙醇中稀释。这是一个常见的错误,经常被遗忘并导致准备失败。另外,不要忘记确保拧紧盖子以防止乙醇蒸发。您的下一个质粒制备将感谢您。
请勿:用微波炉缓冲
某些缓冲液(P2和结合缓冲液)在运输过程中可能会沉淀。但是,请勿尝试使缓冲液微波化,以将沉淀物哄骗回溶液中。取而代之的是,将缓冲液在30-37°C下孵育10分钟,然后颠倒瓶子数次进行混合。
做:检查纯度
下游转化困境的常见原因是盐和蛋白质的污染。每次纯化后,请确保测量A 260 / A 280和A 260 / A 230的比率,以确保您的准备工作干净整洁。通常认为高于1.8的比率是纯净的,没有污染物。
不要:一次处理太多样本
虽然可能很想将制备物堆积起来以最大化时间效率,但质粒纯化中的某些步骤对时间敏感。不要尝试一次处理太多的准备,否则某些准备可能会坐得太久而毁掉。

做:热洗脱
如果质粒> 10 kb,请考虑使用加热至50°C的洗脱缓冲液,以提高柱基质的洗脱效率。另外,在离心之前,洗脱缓冲液可以在色谱柱上放置5-10分钟。
不要:忘记内毒素
一些下游应用,例如敏感细胞系的转染,需要无内毒素的质粒制备物。简单快速的内毒素去除技术可以快速消除这些污染物,从而确保了高质量的转染级质粒。
可以:完美的质粒纯化
虽然清单可以继续,但这些是质粒纯化中最重要的“注意事项”。尝试遵循它们进行下一次纯化,并确保立即成功地获得干净,纯净的质粒制备物的高产量!
(本文转自zymo research 本司不对翻译的准确定做保证,请参阅原文!)